THE UNDERLYING CAUSE OF
MYELOFIBROSIS MAY BE FOUND
AT THE EPIGENETIC CORE1
New discoveries suggest an opportunity for disease modification1
Targeting drivers of myelofibrosis (MF) progression at the epigenetic level, as well as JAK/STAT dysregulation, may offer a new opportunity for disease modification beyond the current standard of care.



Discover the potential of targeting BET proteins in MF with Dr Andrew Kuykendall
This is a very heterogeneous disease, and there’s probably multiple different inflammatory pathways that are being utilized within this.
And so while many of the characteristic signs and symptoms and hallmarks of the disease come from activation of JAK-STAT, the complexity probably comes from utilization of other inflammatory pathways.
And that’s where BET inhibition really comes into focus.
We know that JAK inhibitors help to block that pathway, the JAK-STAT pathway, and thereby lead to clinical efficacy from that standpoint; however, we’ve also learned that there’s other pathways that are included.
And oftentimes those may be critical for some of these underserved aspects of the disease that are ineffectively impacted by JAK inhibitors.
One idea has been to target these epigenetic modulators such as BET proteins.
So, BET proteins have many different target genes that they help to regulate, some of which are involved with the NF-kappa-B pathway.
And this NF-kappa-B inflammatory pathway has come into critical focus over the last few years.
This is part of the innate immune system but activation of some of these NF-kappa-B pathway genes has really shown to have clinical impact within patients with myelofibrosis.
Some of the key upregulated genes within myelofibrosis are shown not to be part of the JAK-STAT pathway.
And here we can see that potentially by impacting the NF-kappa-B pathway by targeting BET proteins, we may be able to have more substantial control or impact upon this disease process.
At least that’s what preclinical models would suggest, and certainly further investigation is warranted.
We believe that in preclinical models that by blocking the NF-kappa-B pathway by targeting BET proteins, or supplying BET inhibition, we may be able to achieve control of 4 different hallmarks of this disease.
We’ll be able to improve megakaryocyte differentiation, thereby reducing bone marrow fibrosis.
We may be able to improve effective hematopoiesis and improve anemia, while also being able to downregulate some of these inflammatory cytokines that lead to an increased symptom burden and prolonged splenomegaly.
So, BET inhibition is very exciting in terms of what it can potentially bring to the table for myelofibrosis.
References:
- Al-Ali HK, Griesshammer M, le Coutre P, et al. Safety and efficacy of ruxolitinib in an open-label, multicenter, single-arm phase 3b expanded-access study in patients with myelofibrosis: a snapshot of 1144 patients in the JUMP trial. Haematologica. 2016;101(9):1065-1073. doi:10.3324/haematol.2016.143677
- Mesa R, Su Y, Woolfson A, et al. Development of a symptom assessment in patients with myelofibrosis: qualitative study findings. Health Qual Life Outcomes. 2019;17(1):61. doi:10.1186/s12955-019-1121-1
- Guglielmelli P, Rotunno G, Pacilli A, et al. Prognostic impact of bone marrow fibrosis in primary myelofibrosis. A study of the AGIMM group on 490 patients. Am J Hematol. 2016;91(9):918-922. doi:10.1002/ajh.24442
- Lacout C, Pisani DF, Tulliez M, et al. JAK2V617F expression in murine hematopoietic cells leads to MPD mimicking human PV with secondary myelofibrosis. Blood. 2006;108(5):1652-1660. doi:10.1182/blood-2006-02-002030
- Takenaka K, Shimoda K, Akashi K. Recent advances in the diagnosis and management of primary myelofibrosis. Korean J Intern Med. 2018;33:679-690. doi:/10.3904/kjim.2018.033
- NCCN Clinical Practice Guidelines in Oncology (NCCN Guidelines®) for Myeloproliferative Neoplasms V.3.2023. © National Comprehensive Cancer Network, Inc. 2023. All rights reserved. Accessed October 1, 2023. To view the most recent and complete version of the guideline, go online to NCCN.org
- Harrison CN, Schaap N, Mesa RA. Management of myelofibrosis after ruxolitinib failure. Ann Hematol. 2020;99(6):1177-1191. doi:10.1007/s00277-020-04002-9
- Scherber R, Mesa R. Management of challenging myelofibrosis after JAK inhibitor failure and/or progression. Blood Rev. 2020;42:100716. doi:10.1016/j.blre.2020.100716
- Pemmaraju N, Verstovsek S, Mesa R, et al. Defining disease modification in myelofibrosis in the era of targeted therapy. Cancer. 2022;128:2420-2432. doi:10.1002/cncr.34205
- Wilkins BS, Radia D, Woodley C, Farhi SE, Keohane C, Harrison CN. Resolution of bone marrow fibrosis in a patient receiving JAK1/JAK2 inhibitor treatment with ruxolitinib. Haematologica. 2013;98(12):1872-1876. doi:10.3324/haematol.2013.095109
- Vachhani P, Verstovsek S, Bose P. Disease modification in myelofibrosis: an elusive goal? J Clin Oncol. 2022;40(11):1147-1154. doi:10.1200/JCO.21.02246
- Kleppe M, Koche R, Zou L, et al. Dual targeting of oncogenic activation and inflammatory signaling increases therapeutic efficacy in myeloproliferative neoplasms. Cancer Cell. 2018;33(1):29-43. doi:10.1016/j.ccell.2017.11.009
- Verstovsek S, Mesa RA, Gotlib J, et al. A double-blind, placebo-controlled trial of ruxolitinib for myelofibrosis. N Engl J Med. 2012;366(9):799-807. doi:10.1056/NEJMoa1110557
- Jiang Q, Jamieson C. BET'ing on dual JAK/BET inhibition as a therapeutic strategy for myeloproliferative neoplasms. Cancer Cell. 2018;33(1):3-5. doi:10.1016/j.ccell.2017.12.007
- Ross D, Babon J, Tvorogov D, et al. Persistence of myelofibrosis treated with ruxolitinib: biology and clinical implications. Haematologica. 2011;106(5):1244-1253. doi:10.3324/haematol.2020.262691
- Copher R, Kee A, Gerds A. Treatment patterns, health care resource utilization, and cost in patients with myelofibrosis in the United States. Oncologist. 2022;27(3):228-235. doi:10.1093/oncolo/oyab058
- Naymagon L, Mascarenhas J. Myelofibrosis-related anemia: current and emerging therapeutic strategies. Hemasphere. 2017;1(1):e1. doi:10.1097/HS9.0000000000000001
- Bose P, Verstovsek S. JAK inhibition for the treatment of myelofibrosis: limitations and future perspectives. Hemasphere. 2020;4:4(e424). doi:10.1097/HS9.0000000000000424
- Mascarenhas J, Gerds A, Verstovsek S. Paradigm shift: combination BET and JAK inhibition in myelofibrosis. Leukemia. 2021;35(12):3361-3363. doi:10.1038/s41375-021-01405-z
- Hilton J, Cristea M, Postel-Vinay S, et al. BMS-986158, a small molecule inhibitor of the bromodomain and extraterminal domain proteins, in patients with selected advanced solid tumors: results from phase 1/2a trial. Cancers. 2022;14:4079. doi:10.3390/cancers14174079
The MPN Landmark Survey reports that the delaying or slowing of disease progression is a top treatment goal for both patients and physicians.2
Preventing the progression of MF or even modifying the disease could help fulfill this shared goal.
A need exists for novel, disease-modifying advancements that deliver deep, durable responses in all 4 hallmarks of myelofibrosis1,3,4
While JAK inhibitors address some aspects of the disease, no agent provides broad disease control.3,5-7
Sustained symptom reductions
Resistance to JAK inhibition monotherapy is not uncommon and loss of a clinical response may include resumption or exacerbation of constitutional symptoms, or disease progression.8
Sustained SVR of ≥35%
Patients with MF taking JAK inhibitors who achieve an SVR of ≥35% may stop responding before 1 year.3,9
- Although JAK inhibitors have demonstrated SVRs at Week 24, a majority of patients may not achieve an SVR of ≥35%3,10,11
Sustained improvement in hemoglobin levels
Some JAK inhibitors may exacerbate anemia due to the on-target inhibition of JAK2.4,12
- Addressing the multifactorial nature of anemia remains a key challenge when treating patients with MF4
Restoration of the bone marrow microenvironment
The reversal of the impairment of the bone marrow microenvironment and reductions in bone marrow fibrosis have not been significantly addressed by JAK inhibitor monotherapy.7
BET proteins are emerging as a promising therapeutic target in MF. Inhibition of BET proteins may help normalize hematopoiesis and modify the underlying cause of the disease.1
There are multiple molecular drivers that contribute to the progression of MF.13-15
BET: The BET family of proteins consists of epigenetic modulators involved in gene transcription.1,13
JAK: In MF, mutations in the JAK/STAT signaling pathway in hematopoietic cells lead to increased levels of proinflammatory cytokines.1
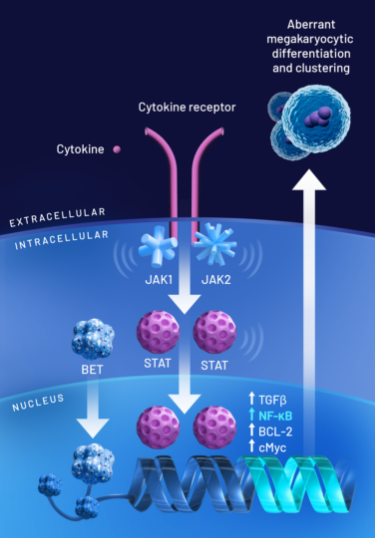
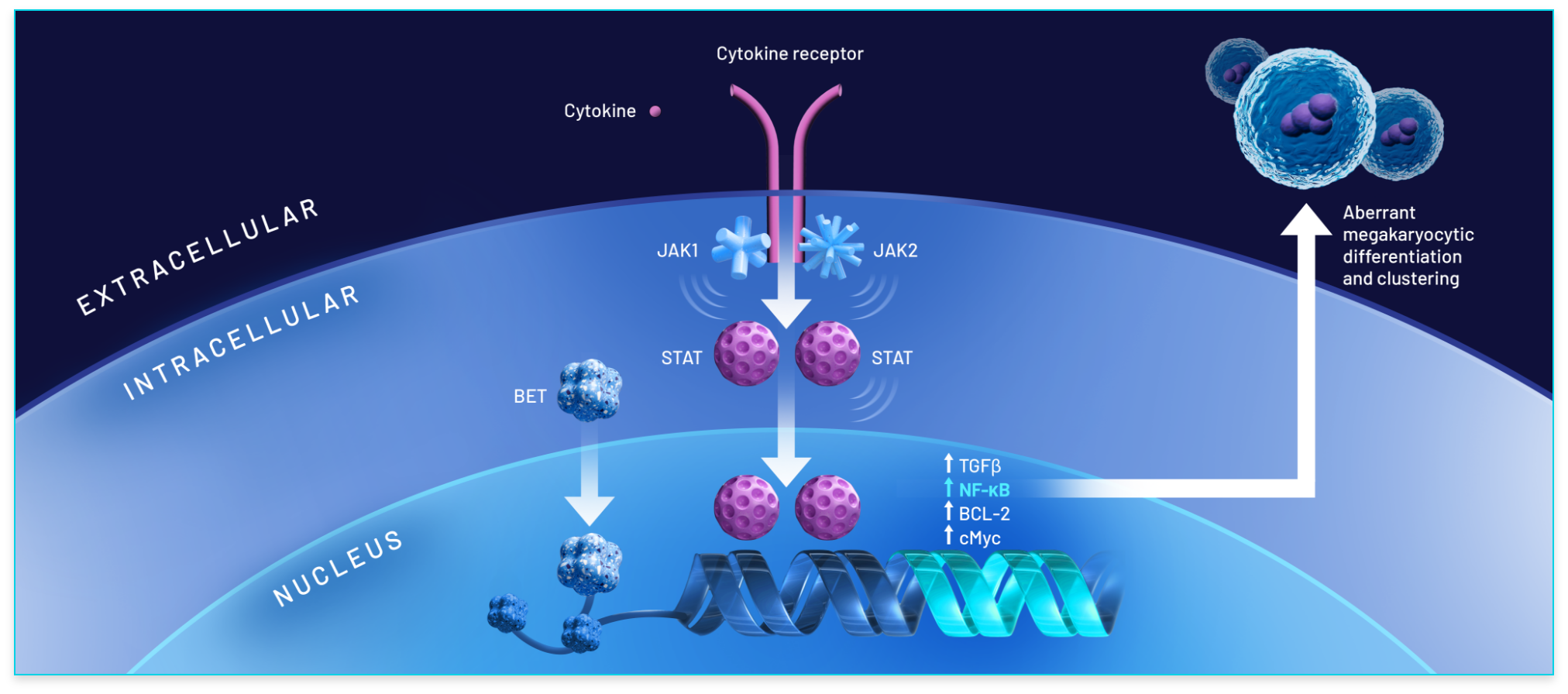
BET proteins play an important role in the progression of MF1,16:
- The BET family of proteins binds to chromatin to modulate the transcription of genes, several of which are involved in cancer13-15
- NF-κB hyperactivation induces the production of proinflammatory cytokines, leading to inflammation and cancer cell development1,16
- In MF, overexpression of BET proteins promotes aberrant megakaryocyte production and leads to subsequent deposits of collagen in the bone marrow, worsening fibrosis and resulting in anemia15,17,18
Dual inhibition of BET protein activity and the JAK/STAT pathway suggests the potential for disease modification1